.svg)
بیماری اس ام ای چیست؟
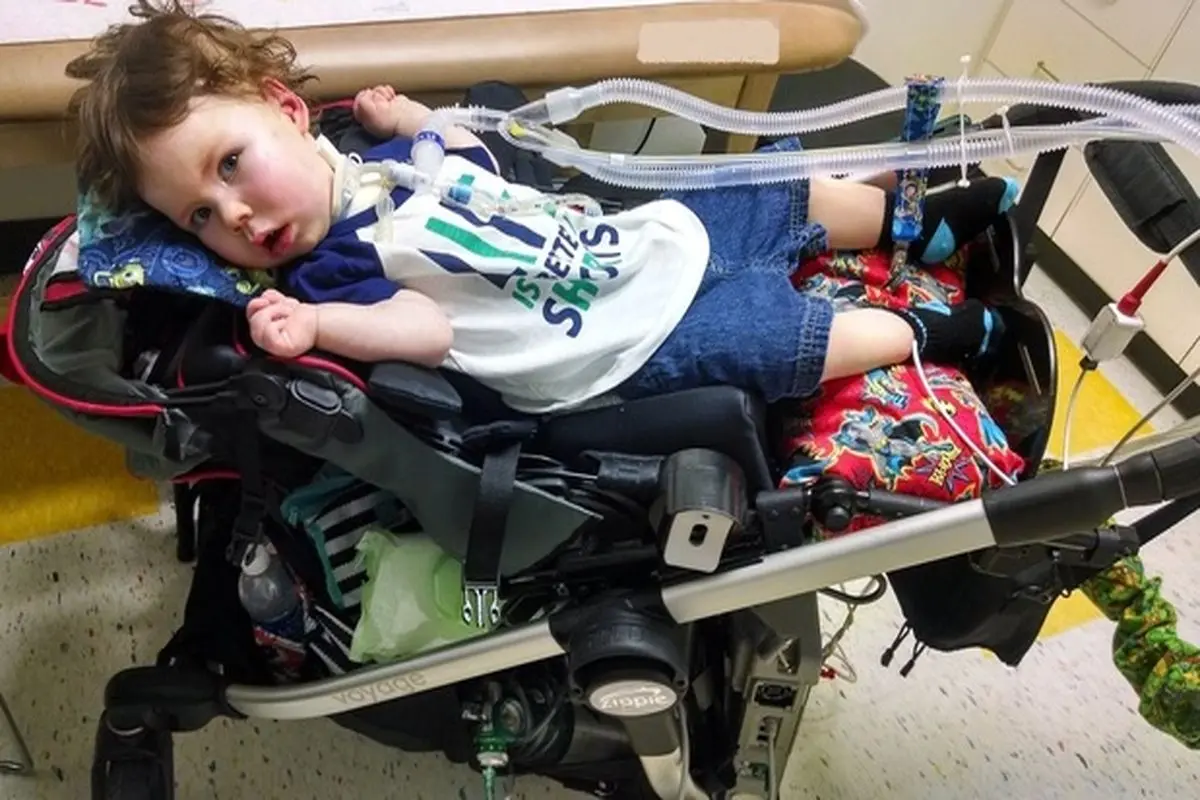
پارسینه: آتروفی عضلانی نخاعی یا همان اس امای (SMA) به تحلیل یا کاهش اندازه عضلات گفته میشود که در نهایت منجر به کم شدن قدرت و تحرک ماهیچهها میشود.
بیماری SMA یا آتروفی عضلانی_نخایی یک اختلال ژنتیکی نادر است که در آن سلولهای عصبی در نخاع دچار مرگ زودرس شده و این امر موجب میشود که ماهیچههایی که به طور معمول توسط آن سلولهای عصبی کنترل میشود، دچار تحلیل رفتگی شوند. در این بیماری بدون در نظر گرفتن نژاد یا جنس از هر ۱۰ هزار نوزاد در سراسر جهان یک نفر از طریق اتوزومی مغلوب تحت تاثیر قرار میگیرد.
نشانهها و علائم بیماری SMA معمولاً در چند ماهه اول بعد از تولد در فرد بیمار قابل مشاهده است که این علائم میتواند تا بزرگسالی ادامه یابد، اما نوزادانی که با شدیدترین شکل SMA متولد میشوند، اغلب به تولد دوم نمیرسند. این بیماری بر تواناییهای ذهنی تاثیر نمیگذارد اما میتواند بر توانایی فرد در انجام عملکردهای اساسی جسمی مانند راه رفتن، تنفس و بلع تاثیر بگذارد.
تشخیص SMA با درک رابطه بین مغز، اعصاب و عضلات
برای درک SMA باید رابطه بین مغز، اعصاب و عضلات را تشخیص دهید. هنگامی که مغز میخواهد ماهیچهای را منقبز کند یک سیگنال از طریق نورون حرکتی فوقانی میفرستد که پیام را از مغز به نخاع میبرد سپس از آنجا از طریق نورون حرکتی تحتانی پیام را از نخاع به محل اتصال عصبی_نخاعی میبرد. نورون حرکتی پایینی که باعث انقباض ارادی ماهیچه اسکلتی میشود نورون حرکتی آلفا نام دارد جسم سلولی این روزها در شاخ قدامی نخاع قرار داشته و آکسونهای آنها نیز از نخاع شروع و تا سلول ماهیچهای ادامه پیدا میکند.
این بیماری که با تحلیل رفتن نورونهای حرکتی ذکر شده و تحلیل پیشرونده قدرت ماهیچهای مشخص میشود به علت کمبود پروتئین smn به وجود میآید. افراد سالم دارای دو ژن تولیدکننده پروتئین smn بر روی کروموزوم q۵ هستند.
ژن smn۱ و کپیهایی از این ژن به نام smn۲. افراد مبتلا به SMA فاقد ژن smn۱ هستند و برای تولید پروتئین smn به یک یا تعداد بیشتری کپی از ژن smn۲ وابسته هستند.
تعداد کپیهای ژن smn۲ در مبتلایان به smn با شدت بیماری آنها ارتباط دارد، اما این ژن پشتیبان ضعیفی است. مطالعات آزمایشگاهی نشان داده است که ژن smn۲ فقط در ۱۰ درصد از موارد به پروتئین smn کامل و کارآمد تبدیل میشود. این اتفاق از یک اشتباه در هنگام ویرایش RNA ناشی میشود.
این بیماری ۴ نوع اصلی داشته که هر نوع آن با توجه به شدت و اثر متفاوتی که بر هر فرد میگذارد، تشخیص داده میشود. تقریباً نیمی از جمعیت مبتلایان این بیماری را نوع اول آن تشکیل میدهند که علائمها تا قبل از شش ماهگی بروز پیدا میکند و این شدیدترین نوع SMA است. این بیماران هیچگاه قادر به نشستن، بدون وابستگی به دیگران نبوده و انتظار میرود طول عمر کمتر از دو سال داشته باشند.
در مبتلایان به نوع دوم SMA علائم شروع بیماری از ۶ تا ۱۸ ماهگی بروز پیدا میکند و به طور معمول قادر به نشستن، بدون اتکا به دیگران هستند، اما برای ایستادن نیاز به کمک دارند. طول عمر قابل انتظار برای این بیماران بیشتر از دو سال است. یا پیشرفت بیماری در مبتلایان به هر دو نوع ظرف پیش رونده در عضلات بین دندهای باعث ناکارآمدی تنفسی میشود که ادامه این روند ممکن است نارسایی تنفسی نیز ختم شود.
مبتلایان به نوع سوم SMA به طور معمول در مراحلی از زندگی خود، علائم حرکتی اصلی را به صورت خفیف بروز میدهند و ممکن است بتوانند در بزرگسالی به طور مستقل راه بروند. طول عمر قابل انتظار برای این بیماران قابل قیاس با افراد سالم است. با بروز دیر هنگام علائم، ضعف پیشرونده پاها در بیماران میتواند باعث از دست دادن کامل توان حرکتی و نیاز به استفاده از وسایل حرکتی شود، این نقص حرکتی میتواند باعث چاقی و ایجاد بیماریهای ارتوپدی از جمله انحنای ستون مهره شود که همگی این عوارض با کاهش تحرک در مبتلایان در ارتباط هستند.
نیاز سلول ها به پروتئین برای کارکرد ماهیچه ها
برای کارکرد ماهیچهها در بدن، سلولهای ما به پروتئین نیاز دارند. در بدن یک فرد سالم، ژنها قادر هستند پروتئینهای لازم را در زمان و مقدار مناسب تولید کنند. یکی از پروتئینهایی که برای عملکرد سلول بسیار مهم است smn نامیده میشود. در نوعی اختلال ژنتیکی به نام آتروفی عضلانی_ نخاعی نوعی یک، سلولها به دلیل داشتن ژنهای معیوب نمیتوانند به اندازه کافی پروتئین smn سالم تولید کنند.
در این بیماری سلولهای عصبی که در آنها فرآیند تولید پروتئینهای smn به خوبی انجام نمیگیرد، میمیرند و هنگام مرگ سلول بیمار توانایی استفاده از عضلات خود را از دست میدهد به صورتی که کودک مبتلا به SMA یک ماه پس از تولد با ضعف و تحلیل رفتن عضلات خود مواجه میشود.
ضعف و تحلیل رفتن عضلات پس از تولد کودک باعث میشود عملکردهای اساسی مانند تنفس، غذا خوردن و خوابیدن به خطر بیفتد. در برخی از آزمایشهای بالینی محققان از ژن درمانی برای انتقال ژن سالم smn۱ به سلولهای بیمار استفاده میکنند.
ZolgenSMA یک دارو برای درمان بیماری SMA بوده که در سطح ژن عمل میکند. این دارو حاوی نسخه سالم از ژن smn انسانی بوده و جایگزین ژن smn میشود که یا در بیماران وجود ندارد و یا فاقد عملکرد مناسب است. این نسخه سالم از ژن smn در یک ویروس قرار میگیرد و این گونه عمل میکند که ژن را به صورت ایمن یعنی حالتی که ویروس تنها حاوی ژن smn باشد را به محل قرارگیری نورونهای حرکتی بدن منتقل میکند.
هنگام تزریق ماده، ویروسها ژن سالم را به سلولهای بدن منتقل و مهمتر از همه اینها از سد خونی مغزی نورونهای حرکتی در نخاع، عبور میکند و هنگامی که وکتور حامل ژن مورد نظر به مقصد خود میرسد، ژن سالم smn۱ به سلول منتقل میشود اما با DNA اقدام نشده و در عملکرد ژنهای دیگر اختلال ایجاد نمیکند.
اگرچه این دو عملکرد نورونهای حرکتی از دست رفتن نمیتواند برگرداند اما میتواند عملکرد نورونهای حرکتی که تاکنون دچار مرگ نشده اند را حفظ کنند و حداکثر طول عمر کودکان مبتلا به SMA را افزایش دهد. هنگامی که ویروس ژن smn۱ را به سلول میرساند، متلاشی میشود و در نهایت با ورود ژن سالم، سلول میتوان پروتئین smn با عملکرد طبیعی ایجاد کند.
قدم دوم در پروسه ایجاد داروی جدید فاز توسعه پیش بالینی است که در آن به تحقیق و جمع آوری تمامی اطلاعاتی پرداخته میشود که کمیت دارو را تضمین میکند. این مرحله معمولا شامل تستهایی است که به صورت in Vivo انجام میشود که در آن از مدل های حیوانی استفاده میشود در مقابل آن in vitro قرار دارد که نیازی به استفاده از هیچ میکرواورگانیسم زندهای ندارد. امروزه انواعی از عاملین AAV وجود دارد که از طریق تحقیقهای in Vivo و in vitro کشف و توسعه یافتند تا امنیت و تاثیر دارو را در هنگام درمان موقعیتهای پزشکی گوناگون ثابت کند.
کارآزماییهای موفقیت آمیز در زمینه درمان AAV
اکنون بر ۳ تا از کارآزماییهای موفقیت آمیز در زمینه درمان AAV میپردازیم. مورد اول یک مورد in Vivo را نشان میدهد که در آن یک حامل AAV خاص با موفقیت به مجرای تنفسی خرگوشها تحویل و به طور طبیعی بروز داده شد و نشان داد که این روش میتواند یک درمان ایمن و بالقوه بر روی بیماران فیبروکیستیک بزرگسالان باشد.
آزمایش دوم in Vivo نشان میدهد، یک حامل ویژه AAV قادر به انتقال ژن حیاطی به عضله اسکلتی و سیستم عصبی مرکزی است که به بدن اجازه میدهد انواع مختلف پروتئین را تولید کند و در مورد آخر از یک حامل ویژه AAV برای افزایش تولید دوپامین در بیماران مبتلا به پارکینسون استفاده شد، زیرا افزایش تولید دوپامین ممکن است با کاهش علائم بیماری مرتبط باشد.
سومین مرحله در فرآیند کشف دارو چالش برانگیزترین و عمیقترین مرحله آن است. در این مرحله از توسعه تحقیقات بالینی گستردهای برای آزمایش اثر بخشی و عوارض جانبی بالقوه دارو انجام میشود. تیمی از محققان یک کارآزماینی بالینی را ایجاد کردند که به کشف درمان بیماری SMA منجر شد. آنها با تزریق وریدی ناقل ژن AAV در نوزادان با آتروفی عضلانی شدید شاهد بهبود عملکرد ضعیف نورونهای حرکتی بودند، پس از آن ۳ آزمایش بالینی مختلف بر روی بیماران مبتلا به SMA نوع یک انجام شد.
این آزمایش بر روی ۱۵ بیمار SMA نوع یک انجام شد که از این ۱۵ بیمار، ۱۲ بیمار دوز بالایی از زوجنزما و ۳ بیمار دوز کمی را دریافت کردند. نتایج این آزمایش نشان داد که ۱۲ بیمار گروه اول بدون هیچ پشتیبانی قادر به تنفس طبیعی بودند و تنها حرکت عضلانی در آنها بهبود یافت بلکه قادر به نشستن و حتی راه رفتن نیز شدند. ۳ بیمار که دوز کم را دریافت کردند به پشتیبانی تنفسی کامل نیاز داشتند. هدف این آزمایشات انتقال ژن سالم به سلولها و تولید مقدار کافی از پروئتینهایی است که به درستی کار میکند.




































ارسال نظر